Contents
Adhering to industry-specific rules and regulations is an essential prerequisite for any product. Medical device regulations for PCBA are crucial to satisfy acceptance criteria for various medtech equipment.
The devices that are heavily reliant on populated boards include:
- Blood pressure monitor
- Heart monitors
- Pacemakers
- Body temperature monitor (thermometer)
- Cochlear implants
- Imaging and diagnostic instrument
- EMG (electromyography) activity system
- Neurostimulator
- Handheld monitors
These devices should function precisely for the right diagnosis and treatment of the patients. This requires a high level of inspection since it is a matter of human lives. Several bodies across the globe have issued guidelines for such instruments.
What are the medical device regulations for PCBA?
Authorities like IPC, ISO, and FDA (Food and drug administration) have released some standards for populated circuit boards used in medical equipment. These guidelines include all the aspects from production to approval. It is mandatory to follow all these regulations during medical PCB assembly to ensure reliability.
IPC
IPC is a body that regulates the assembly and production standards of electronic components and assemblies. There are several important standards:
- IPC-A-600: This standard specifies the acceptability of circuit boards.
- IPC-A-6012: It describes both the qualification and performance requirements for PCBs.
- IPC-A-610: It addresses the acceptability of electronic assemblies. The IPC has different standards for wire harnesses, soldering, enclosures, and cables.
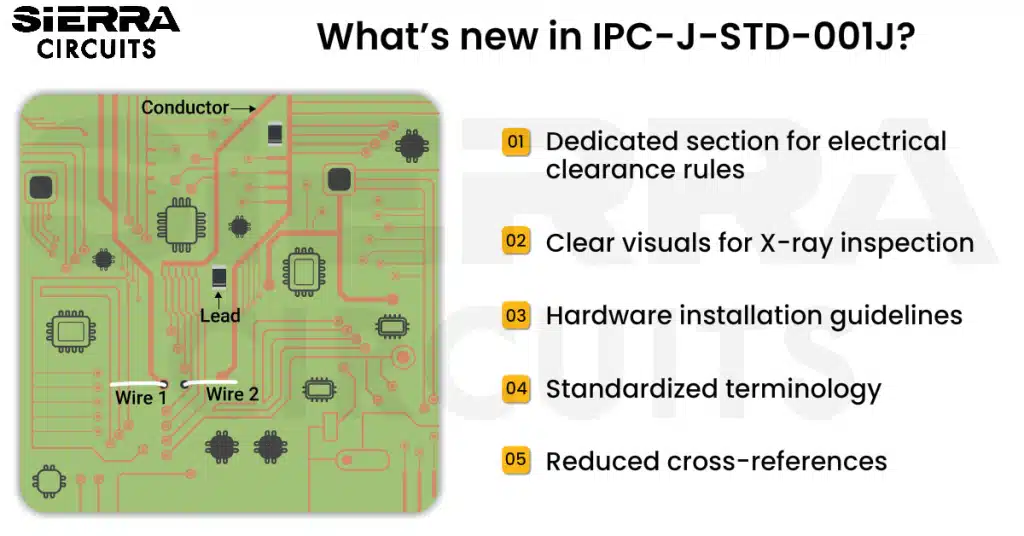
To learn more about material specifications and board acceptability criteria, see IPC J-STD-001 standard soldering requirements.
PCB Design for Assembly Handbook
6 Chapters - 50 Pages - 70 Minute ReadWhat's Inside:
- Recommended layout for components
- Common PCB assembly defects
- Factors that impact the cost of the PCB assembly, including:
- Component packages
- Board assembly volumes
Download Now
ISO
ISO is the acronym for international organization for standardization. It publishes various standards related to the design, manufacture, and testing of circuit boards.
The ISO 9000 standard is mandatory for OEMs to certify the safety of medical equipment and its function. ISO 13485:2016 is an updated quality management system for medtech devices that focuses on current industry practices. In this new version, there is a strong focus on risk management and risk-based decision-making. It also includes enhancements related to regulatory concerns.
FDA
The FDA’s center for devices and radiological health (CDRH) is responsible for imposing laws on companies that manufacture, re-label, re-package, and import medical devices into the United States. Moreover, CDRH monitors electronic products that emit radiations such as lasers, X-ray systems, ultrasound devices, etc.
FDA categorizes medical devices into three classes: class 1, class 2 and class 3.
- Class 1 devices include adhesive bandages, sunglasses, IV (intravenous therapy) stand, etc. These are generally low-risk instruments.
- Class 2 comprises intermediate-risk instruments like syringes, powered wheelchairs, surgical masks, etc.
- Class 3 consists of high-risk devices such as heart valves, implantable neuromuscular stimulators, etc. This category is the most stringent.
There are several aspects for FDA compliance as follows:
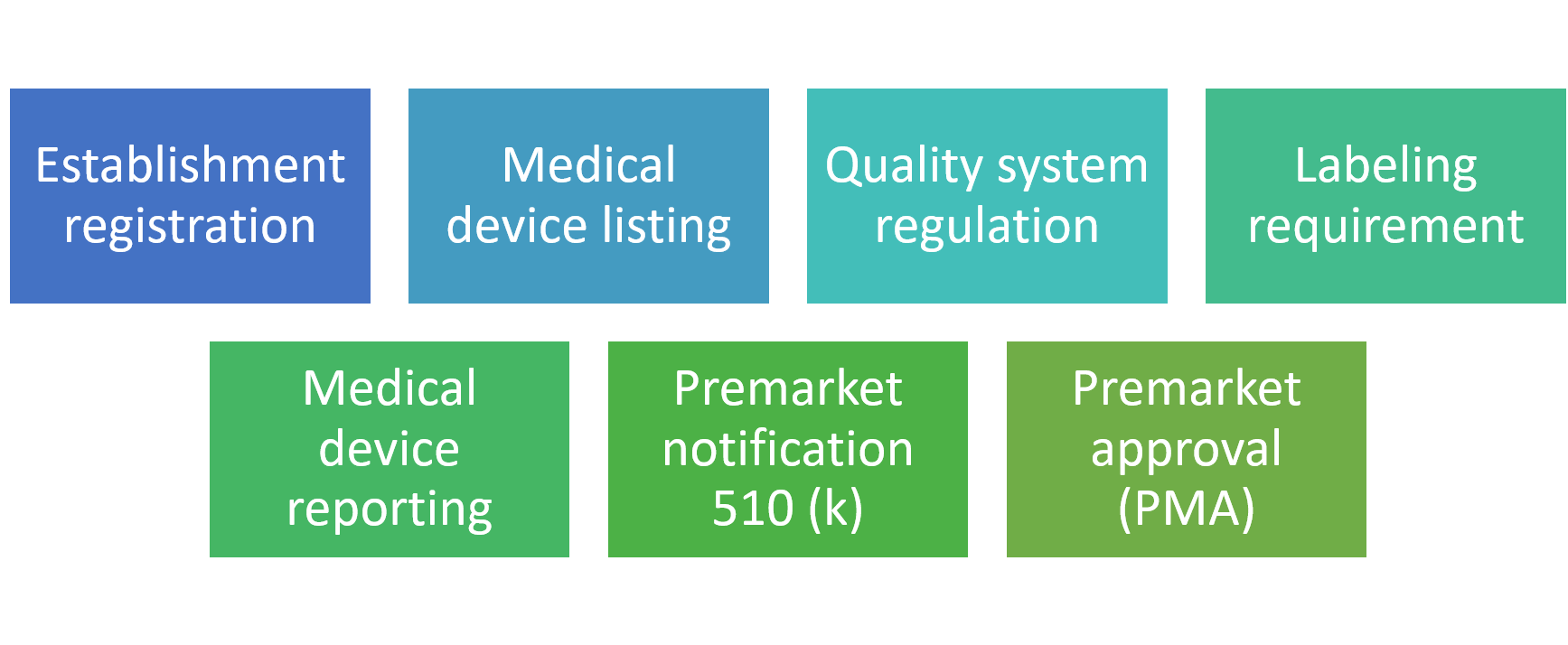
Establishment registration
It is crucial for domestic and foreign manufacturers to register their establishments with the FDA. Establishments need to register electronically unless a waiver has been allowed by FDA. It is mandatory to complete the annual verification of this information between 1st October and 31st December each year. Manufacturers from overseas must also appoint a U.S. representative.
Medical device listing
Another requirement that manufacturers must comply with is the device listing. The following elements should be listed:
- Contract manufacturers (CM)
- Manufacturers
- Contract sterilizers
- Relabelers and repackagers
- Remanufacturer
- Specification developers
- Manufacturers of accessories and components sold directly to the end-user
- Reprocessors single-use devices
- U.S. manufacturers of export only devices
Read our case study: meeting the robotic surgery PCB challenges to learn how we navigated signal integrity, thermal, and space constraints to deliver a high-performance PCB that meets medical-grade reliability.

Quality system regulation
Quality system regulation defines requirements related to designing, procuring, production, packaging, labeling, storing, fitting, and maintaining medical devices. For ensuring compliance with the QS standards, manufacturing facilities are subject to FDA inspections.
Labeling requirement
Labeling includes the physical label of the device, a description, and informational text that accompanies the devices.
Medical device reporting (MDR)
A key objective of the MDR regulation is to identify serious adverse events related to medical devices and to monitor them. It is necessary to notify FDA about the situations that may have caused serious injuries or even death. Similarly, any malfunctions should be reported. The regulation aims to discover and fix problems as early as possible.
Premarket notification 510 (k)
The law, medical device user fee and modernization act allows FDA to charge compensation for medical device premarket notification 510 (k) reviews. Some small businesses may be eligible for a reduced cost.
It is not commercially possible to distribute a device requiring 510(k) submission until the FDA demonstrates valid equivalence.
Premarket approval (PMA)
Class III products need PMAs because they possess significant risk of illness, injury, or cannot be compared directly to class I and II products. Premarket approval is a complex process that includes the submission of clinical data to support claims made about the device.
Sierra Circuits is not an OEM, hence FDA certification is not applicable.

What is QMS in medical devices?
QMS is a quality management system that defines processes, policies, and procedures essential for the planning and execution of a particular product. It focuses on customer demands and enhances their satisfaction level. For medical devices, ISO has issued a standard 13485:2016. This standard includes the key business policies, processes, forms, and instructions. Additionally, it includes their sequence, interactions, and resources for conducting business with a medical device manufacturer.
Documentation is a vital element in QMS. Some of the main documents include:
- Quality manual
- Medical device file
- Document control
- Control of records
- Quality policy
- Additional key docs such as files, formats, templates, etc.
At Sierra Circuits, implantable class 3 medical devices are excluded from ISO 13485:2016 certification.

Need help ensuring your medical device PCB assemblies meet regulatory standards? Our engineering team can assist you with FDA/ISO compliance, documentation, risk management, and validation testing to achieve certification and reliability.
You can book a meeting with our experts or call us at +1 (800) 763-7503.

The DMAIC model
Ideally, QMS follows a DMAIC model for any product. This model is the same for populated boards since it covers all the required aspects. Let’s explore this for medical devices:
Define
- Define the scope of risk management activities; explain the medical device and its proposed use.
- Decide roles, responsibilities, and authorities.
- Determine the phases involved in the life cycle.
- Establish guidelines for risk management.
- Assess risk acceptability and risk verification criteria.
- Analyze the risk management activities related to the production and post-production process.
Measure
- Identifications of input and output of the process.
- Determination of data, process map, data collection, etc.
- Specification for devices.
- Measurement of several parameters using scatter plots, pareto charts, etc.
Analyze
- Analysis of collected data to mitigate quality issues and identification of potential defect causes.
- Map and explore possible solutions to overcome problems associated with customers and business.

Improve
- Elimination of various problems.
- Focus on process improvement.
Control
- Documentation control.
- Organizing and standardizing the working process.
There are specific rules for medical products as follows:
- Ensuring the planning, execution, monitoring, and control of medical devices
- Cleaning of product requires process validation and documentation
- Installation and services requirements with proper documentation
- Sterilization of the devices
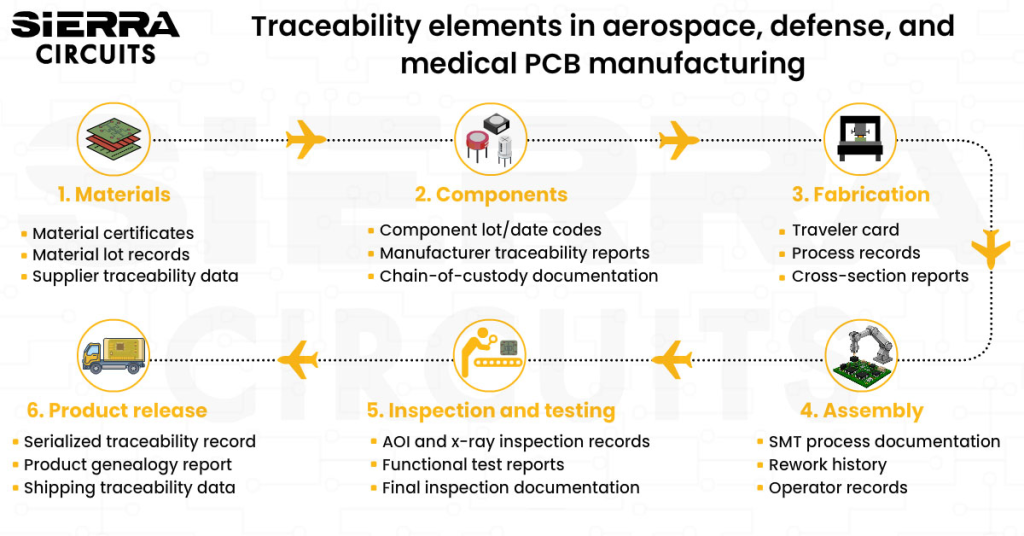
- Validation of process along with traceability
However, sterilization is excluded from Sierra Circuits ISO 13485:2016 certification since its assembly is not the final touch.
To understand the design issues that can impact healthcare device reliability, 7 medical PCB design mistakes that lead to device failure.
HIPAA regulation
HIPAA (Health insurance portability and accountability act) is a federal law that establishes standards for the protection of the health records of individuals. It also includes rules for clearinghouses, health care plans, and various providers of electronic health records. Under this directive, some measures should be taken to protect the privacy of personal health information. There are limits on the uses and disclosures of such information without patient consent. The patient may also request corrections to their health records, see, and obtain copies of them.
Considerations for populated medical PCBs
Manufacturing guidelines for medical equipment and devices are strict and particular. These technologies must adhere to the same high standards when it comes to PCB assembly. When designing and assembling circuit boards for medical devices, consider the following factors:

Safety
Safety is a vital concern in medical devices. These devices are often implanted or directly used on the patient, making circuitry failure a potentially life-threatening hazard. Devices and components must be designed to protect the safety of the patient, regardless of liquid, shock, or temperature extremes. It is necessary to take measures against any potential safety risk.
Regulatory compliance
For the proper functioning of medical devices, it is crucial to follow compliance norms. Regulatory standards are the basis for qualifying the instruments for operation and use. As component tracking is of considerable importance in the medical field, qualified board manufacturers should provide data on the testing and inspection procedures involved in the assembly process.
It is crucial to conduct the inspection tests and document the results in a PCB assembly inspection report. This ensures the quality and reliability of circuit boards.

Reliability
A well-designed circuit board is a crucial factor in creating more predictable and reliable medical equipment and devices. Both attributes must exist for the device to function correctly and deliver consistent results.
Accessibility
There should be an optimal balance between functionality and usability to create a desirable medical product. Medical devices often have specialized forms and properties that make them convenient and practical. It is critical to choose a circuit board manufacturer who will accommodate these design concerns to guarantee maximum value for the device.
Cleanliness
The circuit boards of electronic devices are crucial for their operation, so their functionality must be flawless. The remnants left behind during the manufacturing and soldering processes increase the chances of corrosion and possible failure without any warning. These boards should be thoroughly cleaned to function as intended.
It is important to understand failure modes and their effects to avoid circuit board failures. For a better understanding, check out these 6 types of electronic component failures in PCBs.
Production timeline
The development and introduction of a medical device into the market is vital for its profitability. Electronic medical devices must be manufactured and tested promptly to meet the product development timeline. It is essential to meet tight deadlines to reduce the time-to-market.
To know more about design, check our blog on medtech PCB design considerations with IPC and UL standards.
Sierra Circuits adheres to all aspects including defect identification, resolution, evaluation, traceability, and auditing. In short, it manages the entire lifecycle of printed circuit boards for conforming to all the necessary regulations. We provide efficient boards along with proper documentation for medical applications.
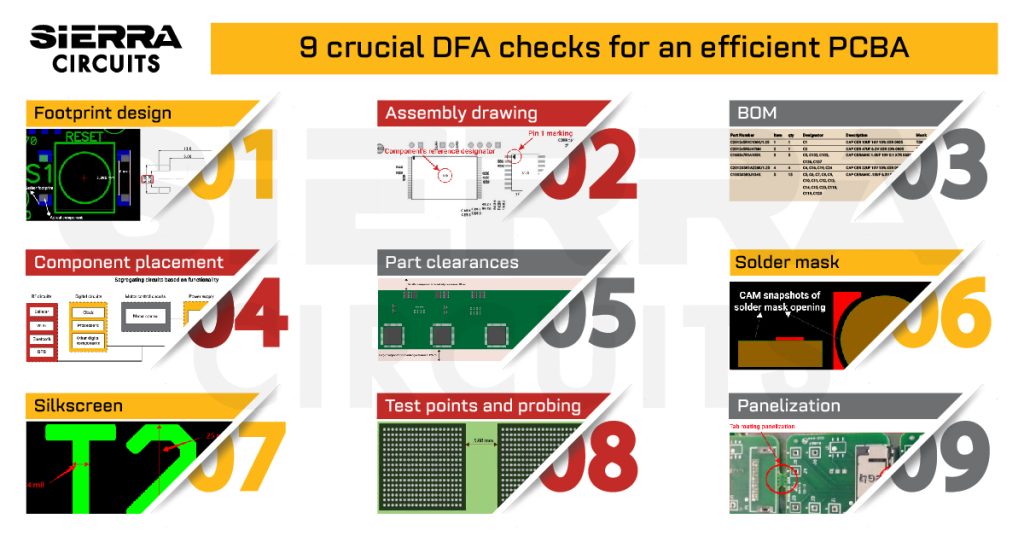
If you have any further questions about medical device regulations for PCBA, please let us know in the comments section. To understand the aspects of design for manufacturability (DFM), download our design guide.

Design for Manufacturing Handbook
11 Chapters - 96 Pages - 90 Minute ReadWhat's Inside:
- Annular rings: avoid drill breakouts
- Vias: optimize your design
- Trace width and space: follow the best practices
- Solder mask and silkscreen: get the must-knows
Download Now